Cardiac Amyloidosis: An In-Depth Analysis
Cardiac amyloidosis is a progressive and life-threatening disorder characterised by the deposition of misfolded amyloid fibrils within the myocardium. This pathological protein accumulation results in increased myocardial stiffness, diastolic dysfunction, and, ultimately, heart failure. Due to its nonspecific clinical manifestations, cardiac amyloidosis often remains undiagnosed until advanced stages, highlighting the necessity for heightened clinical awareness and improved diagnostic methodologies.
 |
| Image source Google |
Aetiology and Risk Factors
Cardiac amyloidosis primarily manifests in two distinct forms:
-
Light Chain (AL) Amyloidosis: Originates from clonal plasma cell dyscrasia, wherein monoclonal light chains misfold and aggregate as amyloid fibrils in various organs, including the heart.
-
Transthyretin (ATTR) Amyloidosis: Arises due to misfolding of the transthyretin (TTR) protein. This can occur as a result of hereditary mutations (hATTR) or age-related destabilisation of wild-type TTR (wtATTR).
Key risk factors include genetic predisposition, chronic inflammatory conditions, multiple myeloma, and advancing age. Epidemiological studies indicate that wtATTR amyloidosis is particularly prevalent in elderly populations, whereas specific TTR mutations disproportionately affect individuals of African and Portuguese descent.
Pathophysiology
The pathogenesis of cardiac amyloidosis involves extracellular deposition of amyloid fibrils within myocardial tissue, disrupting normal cardiac architecture and function. These fibrillar aggregates impair cardiomyocyte contractility, diminish ventricular compliance, and lead to restrictive cardiomyopathy. Furthermore, amyloid deposition interferes with electrical conduction pathways, predisposing patients to arrhythmias and conduction abnormalities. Over time, these pathophysiological changes culminate in progressive heart failure with preserved ejection fraction (HFpEF).
 |
| Image source Google |
Clinical Presentation
The symptomatology of cardiac amyloidosis is highly variable and often mimics other cardiovascular diseases. Key clinical features include:
-
Exercise intolerance and exertional dyspnoea
-
Peripheral oedema and ascites due to right heart failure
-
Orthostatic hypotension resulting from autonomic dysfunction
-
Atrial fibrillation and conduction disturbances
-
Carpal tunnel syndrome, particularly in ATTR amyloidosis
-
Unintentional weight loss and fatigue
Diagnostic Approach
Accurate and timely diagnosis of cardiac amyloidosis necessitates a multimodal approach incorporating imaging, biomarker analysis, and histological confirmation:
-
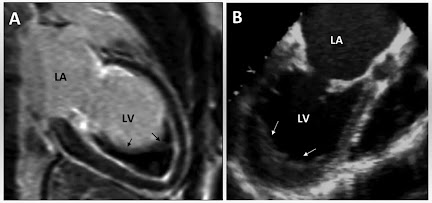
Echocardiography: Demonstrates ventricular hypertrophy with preserved ejection fraction, apical sparing on strain imaging, and restrictive physiology.
-
Cardiac Magnetic Resonance (CMR) Imaging: Utilised for tissue characterisation, revealing late gadolinium enhancement with a global subendocardial pattern indicative of amyloid infiltration.
-
Nuclear Scintigraphy: Differentiates ATTR from AL amyloidosis, with technetium-labelled bone tracers showing avidity for ATTR deposits.
-
Serum and Urine Free Light Chain Assays: Essential for ruling out AL amyloidosis.
-
Endomyocardial Biopsy: Considered the gold standard, with Congo red staining demonstrating apple-green birefringence under polarised light.
-
Genetic Testing: Recommended for suspected hereditary ATTR amyloidosis to guide familial risk assessment and management.
Therapeutic Strategies
Treatment modalities for cardiac amyloidosis are contingent upon the underlying aetiology:
-
AL Amyloidosis:
-
Chemotherapy (e.g., bortezomib, daratumumab) targets clonal plasma cells.
-
Autologous stem cell transplantation (ASCT) may be considered in select patients.
-
-
ATTR Amyloidosis:
-
TTR stabilisers (e.g., tafamidis) prevent protein misfolding and slow disease progression.
-
Gene silencing therapies (e.g., patisiran, inotersen) reduce hepatic TTR synthesis.
-
Liver transplantation remains an option for hereditary ATTR amyloidosis.
-
-
Supportive Measures:
-
Diuretics for volume management
-
Pacemaker implantation for conduction disturbances
-
Exercise and dietary modifications for symptom control
-
Prevention and Risk Reduction
While genetic predisposition renders some cases unavoidable, early intervention and lifestyle modifications may mitigate disease progression. Regular cardiovascular screening in at-risk populations facilitates early diagnosis, and emerging pharmacotherapies offer hope for disease-modifying interventions.
Disease Complications
Without appropriate treatment, cardiac amyloidosis precipitates severe complications, including:
-
Progressive heart failure and cardiogenic shock
-
Ventricular arrhythmias and sudden cardiac death
-
Systemic thromboembolism and cerebrovascular events
-
Multi-organ involvement, particularly renal and hepatic dysfunction
Prognosis
The prognosis of cardiac amyloidosis varies based on the underlying subtype and treatment response. AL amyloidosis, if untreated, has a particularly poor prognosis with a median survival of approximately six months post-diagnosis. However, early intervention with chemotherapy or stem cell transplantation can significantly improve outcomes. ATTR amyloidosis, especially wtATTR, progresses more slowly, with survival often exceeding a decade when managed effectively with disease-modifying therapies such as tafamidis. Early diagnosis and appropriate management are critical in enhancing patient survival and quality of life.
Epidemiological Trends
Recent advances in diagnostic techniques have led to increased recognition of cardiac amyloidosis. AL amyloidosis primarily affects individuals in their 60s, whereas wtATTR amyloidosis is increasingly diagnosed in patients over 70 years of age. With improved awareness and genetic screening, hereditary forms of ATTR amyloidosis are being detected at earlier stages, allowing for timely therapeutic intervention.
Recent Advances in Research
Ongoing clinical trials are exploring novel therapeutic strategies aimed at halting or reversing amyloid deposition. Gene-editing technologies such as CRISPR hold promise for directly modifying pathogenic TTR mutations, while small-molecule inhibitors are being investigated for their role in disrupting amyloid fibril aggregation. Advances in biomarker research are also facilitating early-stage detection, improving prognostic outcomes.
Case Studies
Case Study 1: AL Amyloidosis
A 62-year-old male presented with progressive dyspnoea and neuropathy. Echocardiography demonstrated increased left ventricular wall thickness, and serum free light chain assays confirmed AL amyloidosis. Chemotherapy with bortezomib led to significant haematological response and symptom stabilisation.
Case Study 2: ATTR Amyloidosis
A 70-year-old female with bilateral carpal tunnel syndrome and autonomic dysfunction was diagnosed with ATTR amyloidosis via nuclear scintigraphy. Genetic testing revealed a pathogenic TTR mutation. Tafamidis therapy was initiated, leading to symptomatic improvement and stabilisation of disease progression.
Conclusion
Cardiac amyloidosis is an increasingly recognised cause of heart failure with preserved ejection fraction. Advances in diagnostic imaging, biomarker assays, and targeted therapies have transformed disease management, offering patients improved survival and quality of life. Continued research into disease pathophysiology and novel therapeutics remains crucial for optimising patient outcomes.
References
-
Wechalekar, A. D., et al. (2016). "Systemic amyloidosis." The Lancet, 387(10038), 2641-2654.
-
Gillmore, J. D., et al. (2018). "Nonbiopsy diagnosis of cardiac transthyretin amyloidosis." Circulation, 138(6), 599-609.
-
Maurer, M. S., et al. (2019). "Cardiac amyloidosis: JACC state-of-the-art review." Journal of the American College of Cardiology, 74(22), 2872-2885.
-
Ruberg, F. L., & Berk, J. L. (2020). "Transthyretin (ATTR) amyloidosis: Mechanisms and diagnosis." The Journal of the American College of Cardiology, 75(22), 2846-2861.
-
Hanna, M. (2021). "Novel therapies in cardiac amyloidosis." Circulation: Heart Failure, 14(2), e007973.





No comments:
Post a Comment
Please do not enter any spam link in the comment box